Sindrom Cockayne (sindrom Neill-Dingwall) adalah gangguan multi-sistem resesif autosom yang jarang berlaku yang disebabkan oleh kecacatan molekul yang merosakkan mekanisme pembaikan DNA. Kejadian tahunan di negara-negara Eropah hampir 1 / 200,000
Dengan sindrom Cockayne, sel-sel pesakit menunjukkan kecacatan spesifik pada gen yang terlibat dalam menghilangkan perubahan DNA yang disebabkan oleh UV pada gen yang ditranskripsikan secara aktif. Sekiranya terdapat gejala sindrom ekstra-kulit, kecacatan tambahan dalam transkripsi asas atau pembaikan oksidatif juga diambil kira.
Gejala dan jenis penyakit
Terdapat 3 jenis sindrom Cockayne:
- Jenis I (klasik) - awalnya tidak ada penyimpangan dari norma yang diperhatikan, kadang-kadang mungkin ada mikrosefali, anak mungkin berat badannya bertambah buruk. Secara beransur-ansur, penglihatan dan pendengaran merosot, degenerasi sistem saraf, baik pusat dan periferal, menyebabkan kematian pramatang (dekad pertama - kedua kehidupan)
- Jenis II (sindrom cerebro-eye-facial-skeletal) - yang paling teruk, menyebabkan kematian pada dekad pertama kehidupan. Ia dicirikan oleh gangguan perkembangan neurologi. Tisu lemak dan atrofi otak, katarak dan osteoporosis berkembang. Sindrom COFS adalah bentuk pranatal spektrum klinikal sindrom Cockayne yang dicirikan oleh kongenital minima dan arthrogryposis (kontraktur poliartikular).
- Jenis III - yang paling ringan, gejala serupa dengan jenis I, tetapi kurang teruk. Ini membolehkan anda mencapai usia dewasa, kadang-kadang bahkan 4-5 dekad hidup.
Keterukan gejala dan usia permulaan penyakit berbeza-beza bergantung pada tempat mutasi. Dalam sindrom Cockayne klasik jenis 1, gejala pertama paling kerap muncul pada tahun pertama kehidupan. Kes permulaan awal (usia pranatal) dengan gejala yang lebih teruk (jenis II) dan kes permulaan kemudian dengan gejala yang lebih ringan (jenis III) juga telah dilaporkan.
Gejala penyakit yang paling biasa termasuk:
- perencatan pertumbuhan progresif
- ataksia cerebellar
- kekejangan (pengecutan otot secara umum dan berlebihan yang menghalang pergerakan normal)
- kecacatan intelektual
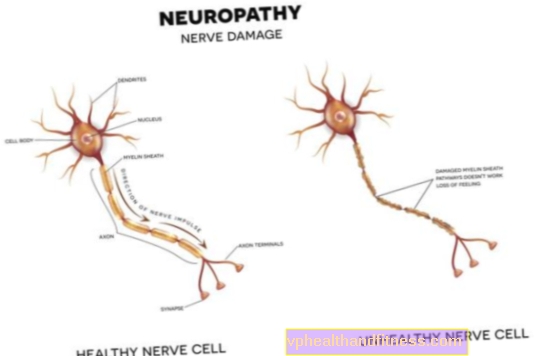
- neuropati sensorineural periferal neuropati
- hilang pendengaran
- retinopati pigmen
- kecacatan gigi (kehadiran karies)
Ciri-ciri wajah yang tipikal termasuk mikrosefali, telinga besar, hidung sempit dan enofthalmia (bola mata runtuh di soket mata apabila kandungan orbit dikurangkan).
Katarak dan kepekaan fotosensitif, serta retinitis pigmentosa, yang dapat menyebabkan kebutaan, telah diperhatikan pada beberapa pesakit.
Disfungsi oklusal dan ginjal, serta kekurangan atau kelewatan pematangan seksual juga diperhatikan.
Risiko mutasi baru dan barah semakin meningkat.
Terdapat kehilangan lemak subkutan yang dapat memberikan penampilan penuaan pramatang pada kulit.
Diagnosis
Penyakit jenis A disebabkan oleh mutasi pada gen ERCC8 pada kromosom 5q11. Jenis B menyebabkan mutasi pada gen ERCC6 pada lokus 10q11.23.
Ia dapat dikenal pasti menggunakan ujian radioaktif dalam kultur fibroblas untuk mengukur pembaikan sintesis DNA selepas sinaran UV. Ujian pembaikan DNA adalah alat penentu untuk diagnosis sindrom.
Diagnosis pranatal
Diagnosis pranatal boleh dilakukan dengan menguji amniosit atau chorionic villi (dengan cara yang sama seperti postnatal) atau secara langsung dengan penjujukan molekul di mana mutasi penyebab penyakit telah dikenal pasti dalam keluarga.
Rawatan
Tidak ada rawatan kausal. Rawatan hanya bersifat simptomatik dan merangkumi terapi fizikal, pelindung sinar matahari, alat bantu pendengaran, dan sering memberi makan tiub atau gastrostomi.







-przyczyny-objawy-leczenie.jpg)





-badanie-poziomu-we-krwi---normy.jpg)